Molecular Dynamics Simulations Reveal Novel 12-Mer Peptide Derived From CPE30 of Clostridium Perfringens Targeting M Cell
Kien-Quang Huynh1,2, Vertika Gautam3, Byeol-Hee Cho4, Yong-Suk Jang4,5, Vannajan Sanghiran Lee3, Hieu Tran-Van1,2*
1 Department of Molecular and Environmental Biotechnology, Faculty of Biology and Biotechnology, University of Science, Ho Chi Minh, Vietnam.
2 Vietnam National University, Ho Chi Minh, Vietnam.
3 Department of Chemistry, Faculty of Science, University of Malaya, Kuala Lumpur 50603, Malaysia.
4 Department of Bioactive Material Sciences, Jeonbuk National University, Jeonju 54896, Souht Korea.
5 Department of Molecular Biology, Jeonbuk National University, Jeonju 54896, South Korea.
* Email: tvhieu @ hcmus.edu.vn; vannajan @ um.edu.my
ABSTRACT
Oral vaccine has been become the most effective strategy in the fight against intestinal infections because of its economy, ease, and capability on inducing responses not only systemic but also local, especially in mucosal compartments. The major obstruction to potent vaccine development is antigen dispersion and tolerance. To overcome this hindrance, we aimed to target M cells, which are the main sentinel gateway for taking up luminal antigens and initiating specific mucosal responses. In this study, 12-mer peptide (CPE12) derived from the C-terminal of 30 amino acids of Clostridium perfringens (CPE30) was predicted to be a good binder using molecular docking and molecular dynamics simulations. To confirm the interaction between CPE12 and M cell receptor, the coding gene for CPE12 was cloned and expressed, CPE12 was then purified, and evaluated ex vivo afterwards. As a result, ex vivo assessment on murine M cells demonstrated that CPE12 had lower binding on murine M cells surface comparing to CPE30. These present results suggest that CPE12 could be a competent candidate peptide for oral vaccine development.
Key words: Claudin-4, CPE, In silico prediction, Oral vaccine, Targeted peptide.
INTRODUCTION
The mucosal immune response plays a vital role in struggling against infections of microorganism. In particular, intestine, the nutrient concentrated body lumen [1, 2], is also the battle frontline where infectious agents start their fight and invasion the most [3]. Unlike mucosal immune response, major isotype secretion in the systemic immune response is serum IgG but not that IgA, thus the prevention of gastrointestinal diseases by injected vaccines does not bring on desirable outcomes because of isotype difference [4]. Therefore, numerous studies were developed to stimulate specific IgA production at infectious sites via M cells targeting.
Microfold cells, also known as M cells, specialized epithelial cells of gut-associated lymphoid tissue (GALT), hold a crucial role in the intestinal immune system [5]. Their unique properties such as lack of microvilli layer, thin glycocalyx, high endocytic activity allow M cells to capture and sample antigens easier than the surroundings and then transport captured antigens from the gut lumen to the underlying layers where mucosal immune system resides [5]. However, the proportion of M cells on the intestinal surface is extremely rare (approximately 5-10% in total epithelial cells), thereby targeting the exact sites to avoid antigen dispersion as well as antigen degradation in the gastrointestinal tract is indispensable for effective mucosal immune response [6]. Rationally, antigens are conjugated to ligands whose cognate receptors express exclusively on M cell surface including lectin, antibody, protein, etc. [7]. Protein emerges amid the receptors as a potential candidate because of its ease for large-scale production. Reportedly, there are proteins and peptides such as FimH [8, 9], Hsp60 [10], Co1 [11, 12], C-terminus Clostridium perfringens enterotoxin (CPE30) [13], etc, which their binding to M cells enhances mucosal immune response. Particularly, peptide or protein-derived peptide is more favorable because its length leaves more room for bulky antigen conjugation without compromising the binding capability in comparison to its full-length protein.
Clostridium perfringens enterotoxin (CPE) is a polypeptide containing 319 amino acids (35.5 kDa). CPE consists of two portions, N-terminus (amino acid 1-200) and C-terminus (amino acid 200-319), in which the latter is a key player binding to its corresponding receptor Claudin-4 on M cells surface [13]. A big body of evidence shows it requires only the last 30 amino acids of CPE (CPE30) to interact effectively with Claudin‑4 instead of full-length CPE [14, 15]. Although the length of CPE30 is quite small, we sought to determine whether there are shorter peptides than CPE30 yet remain binding capacity. To cut the story short, by combining bioinformatics tools such as molecular docking, molecular dynamics based on the X‑ray structure of CPEs and Claudin-4 protein, we discovered that not the whole but a portion of CPE30 is still capable to bind M cell receptor as well as other potential peptides that are outside CPE30. In this study, we revealed a novel peptide comprising of 12 consecutive amino acids inside CPE30, which had binding affinity and molecular energy similar to that of CPE30 and Claudin-4. In order to verify that interaction ex vivo, CPE12 was implemented on murine M cells. This work provided a novel 12-mer peptide for targeting M cells for studies of oral vaccine development.
MATERIALS AND METHODS
Strains and plasmids
The E. coli strain DH5α [F- endA1 hsdR17 (rk-mk-supE44 thi λ-recA1 gyrA96 ∆lacU169 (φ80 lacZ ∆M15)] was used as a host for manipulation of recombinant DNA. E. coli strain BL21 (DE3) (F+ ompT hsdSB (rB- mB-) gal dcm (DE3)) was used for expression of the recombinant protein.
Plasmid pET28a (Addgene, USA) is 5369 bp in size, which is used to express fusion protein CPE12‑GFP and carries the kanamycin resistance gene for screening transformants (Figure 1). GFP was fused to stabilize CPE12 and to facilitate in vivo staining in mouse intestines.
Protein and peptide preparation
The crystal structure of the human Claudin-4 complex with CPE30 with a resolution of 3.5 Å, R‑free value of 0.309, and R-work value of 0.288 was retrieved from protein data bank by PDB ID: 5B2G.[16] Prior to docking, all water molecules were removed from the PDB structures. The linear peptide, CPE12 (GNYPYSILFQKF) was designed based on the C-terminal of CPE30 sequence (SLDAGQYVLVMKANSSYSGNYPYSILFQKF). The structures were prepared for further calculations by reconstructing missing atoms, adding hydrogens, assigning atomic charges and radii from specified force fields using the pdb2pqr software [17] and optimized up to 2000 cycles using CHARM force field. The protein structure was visualized using the Discovery Studio programme 2.5 (BIOVIA Discovery Studio Modelling Environment, San Diego, California, USA).
Protein-protein docking
In parallel, molecular docking studies were carried out using HADDOCK or High Ambiguity Driven protein-protein Docking web server [18]. HADDOCK docking program was based on molecular mechanics (MM) method with three stages of performance that involved rigid-body energy minimization, semi-flexible refinement in torsion angle space, and followed by a final refinement in explicit solvent. The designed CPE12 peptide was docked into the binding site of Claudin-4 where Claudin-4 was treated as a rigid receptor molecule and CPE12 as a flexible peptide. Approximately, 300 docked structures clustered in 10 groups were generated from HADDOCK webserver. The dimension of the grid size was set at 0.5 Å. The HADDOCK score is a combination of 1.0*VDW energy +0.2*ELEC energy +1.0*Desolvation energy. The Z-score indicates how many standard deviations from the average docked structure is in the cluster (the more negative the better binding). The docked conformation from the top cluster with the highest Z-score and binding affinity (∆G) in kcal/mol was selected for further refinement in an aqueous solvent using molecular dynamics (MD) simulations.
Molecular Dynamics Simulations (MDs)
Molecular dynamics simulations were performed on the designed peptide CPE12 and Claudin‑4 complex obtained from docking studies. Residues of Claudin-4 that traverse the membrane were fixed (169-190, 237-259, 278-312, 320-345) during the simulation. MD simulations were performed using the GPU version of Particle Mesh Ewald Molecular Dynamics (PMEMD.CUDA) from AMBER14. At the molecular level, physical forces were implemented using the ff14SB protein force field to carry out MD simulations. Structures were solvated (using tLeap module implemented in AMBER). Water model TIP3P was used, wherein a cubic box of water that extends at least 10 Å from the solute in each direction. A cut-off distance of 15 Å was used to compute the non-bonded interactions (electrostatic interactions and Van der Waals interactions). For each complex, a 100-ns long simulation was performed. To minimize the edge effects, all simulations were performed under periodic boundary conditions, and to treat long-range electrostatics, the particle-mesh-Ewald method was used. The time step of 1fs with a trajectory record over every 0.1ps was set for the simulation. To relax the system prior to MD simulation, the complex (CPE12 and Claudin-4) was minimized using series of steepest descent (SD) and conjugated gradient (CG) under sander module of AMBER14 program (USA). While simulation, the system was heated gradually over a period of 60ps from 0 to 310K (biological temperature), and a force constant of 5 kcal/molÅ2 was applied to fix the protein atoms. A constant pressure of 1 atm for 200 ps (NPT) was applied under Langevin dynamics followed by a 40 ps-volume-constant period (NVT) at a force constant of 2.5 kcal/mol Å2, which was maintained and followed by 100ps dynamics at a force constant of 1.25 kcal/mol Å2. Finally, unrestrained production runs were performed for 100 ns wherein no force was applied on any protein atoms in the NVT ensemble at a constant temperature of 310K. For all analyses, 500 snapshots were taken from the last 5ns of the simulation. To check the system equilibrium, Root-mean-square deviations (RMSD) of all Cα atoms, was performed. Two simulations run for 100 ns with random initial velocity were performed in Data Intensive Computing Centre server (https://www.dicc.um.edu.my).
Construction of plasmid pET28a-cpe12-gfp
The sticky-end strategy was used by adding the restriction sites of BamHI and XhoI in 5’ and 3’ ends, respectively through PCR amplification (Figure 1). PCR was set up in 50 µl volume of MyTaq™ Red DNA Polymerase Kit (Bioline, UK) based on the standard protocol of manufacturer with specific primers containing restriction sequences and plasmid pET28a-gfp was used as the template. Thermo-cycle was set up for cpe12-gfp amplification consisting of an initial denaturation at 95°C for 1 min followed by 30 cycles, each at 95°C for 15 sec, 65.5oC for 10 sec, 72°C for 10 sec, and the final extension at 72°C for 10 min.
Plasmid pET28a was extracted by using the EZ-10 Spin Column Plasmid DNA Minipreps Kit (Biobasic, Canada). After that, plasmid pET28a and amplified gene cpe12-gfp were digested by the same restriction enzymes. Digested cpe12-gfp gene was inserted into digested plasmid pET28a with the ratio of 5:1 using T4 DNA ligase (Thermo scientific, USA), and the ligase product was introduced to E. coli DH5α by the heat shock method. The recombinant plasmid was screened with PCR colony method using T7 primer set. All PCR products were subjected to electrophoresis on 1.5% agarose gel. The plasmids from the positive colony were extracted and the nucleotide sequence of the constructed plasmids was confirmed by sequencing.
Expression of protein CPE12-GFP
Plasmid pET28a-cpe12-gfp was introduced into E. coli BL21 (DE3) by the heat shock method. The positive colony was grown in LB-Kan50 at 37oC and 250 rpm for 16 hours. On the next day, the subculture was inoculated with a ratio of 1:20 until OD600 reached 0.6-1. Next, the protein expression was induced with 0.5 mM IPTG (Biobasic, Canada) and continued to shake at 25oC and 250 rpm for 6 hours. In the next step, the cell pellet was collected, suspended in lysis buffer (20 mM phosphate buffer- 3.8 mM of NaH2PO4, 16.2 mM of Na2HPO4, pH 7.4) and sonicated with ultrasonic cell disruptor. Cell lysis was centrifuged at 13000 rpm for 5 minutes to obtain the supernatant and pellet separately.
Then the supernatant and precipitation of protein were treated with loading buffer 6x and heated at 95oC for 30 minutes; after that, they were subjected to 15% gel SDS‑PAGE with Coomassie staining and Western Blot probed with anti-His-tag antibody (Santa Cruz, USA). GFP protein expression was carried out simultaneously with the same procedure as a control.
Purification of protein CPE12-GFP
The fused protein CPE12-GFP with C-terminus His-tag was purified by IMAC method. The protein sample was prepared then filtered by 0.2 µm membranes. HisTrap HP 5 ml column (GE Healthcare, USA) containing nickel was equilibrated with buffer A (20 mM phosphate, pH 7.4), followed by protein sample loading. The column then washed with buffer A, followed by buffer B (20 mM phosphate, 90 mM imidazole, pH 7.4). Finally, the elution step was carried out with buffer C (20 mM phosphate, 99 mM imidazole). The eluted fractions were assessed by SDS-PAGE with silver staining and analyzed with Gel Analyzer software.
Ex vivo evaluation by M cells of mouse intestines
All mice were maintained in the experimental animal facility and experiments were performed in accordance with the guidelines provided by the Animal Care and Use Committee of the University of Science, VNU-HCM. The Peyer’s patches (PPs) were isolated from the mouse intestine and fixed with 4% paraformaldehyde at 4°C overnight. Fixed PPs were permeabilized with fixation/permeabilization solution (BD Bioscience, USA) as the manufacturer indicated. The PPs were then incubated with recombinant proteins (50 μg) for at 25°C 2 hours followed by incubation with a blocking buffer (PBS with 5% fetal bovine serum and 0.1% glycine). The PPs were also incubated with M cell-specific antibody, NKM 16-2-4 (Miltenyi Biotec Inc., Germany), and finally stained with Alexa fluor 488-conjugated anti-GFP antibody, Alexa fluor 680-conjugated anti-rat IgG antibody and rhodamine-conjugated UEA‑1 (Life Technologies, USA). The specimens were analyzed using super-resolution confocal laser scanning microscopy (LSM 880 with Airyscan, Carl Zeiss, Germany) and the data were analyzed with the ZEN Zeiss program.
RESULTS
Bioinformatic prediction from molecular docking and molecular dynamics
The docking structure showed that the CPE12 bound to the active sites of the Claudin-4 with Z‑score and binding affinity -1.9, -8.5 kcal/mol, respectively showed good affinity. The total protein-peptide interaction energy of the native complex was -691.761 kcal/mol. While overall complex interaction energy within 5 Å residues is -508.718 kcal/mol mainly contributed by their electrostatic and van der Waals interaction energy. In native CPE, there were major contributions of electrostatic and van der Waals energy with values -437.358 kcal/mol and -71.360 kcal/mol, respectively (Supplementary Table S1). The complex structure of designed peptide CPE12 with Claudin-4 obtained from docking studies was subjected to molecular dynamics simulations to study thermodynamics and to analyze the performance of designed peptide based on its binding free energy. The CPE12 and Claudin‑4 complex was found to have the binding free energy in the average of -51.49 ± 0.37 kcal/mol with the energy components; electrostatic, van der Waals and Delta G Gas contributions -68.75 ± 0.89 kcal/mol, -55.92 ± 0.29 kcal/mol and -124.67 ± 0.90 kcal/mol, respectively as illustrated in Table 1. The decomposition energy per residue between Claudin-4 (Residues 1-322) and CPE12 (Residues 323-334) from Run 1 were analyzed using the MM-GBSA method of binding free energy calculation, to study the contribution of each residue in the receptor and ligand interactions. As shown in Figure 2, residues ILE196, VAL197, THR198, VAL211, LYS221, VAL300 belonging to Claudin-4 and TYR335, ILE339, IEU340, PHE341, PHE344 belonging to CPE12 were found to contribute the most to the binding energy with the absolute relative decomposition energy greater than 1 kcal/mol. Analysis of RMSD for Cα atoms was carried out for 100 ns simulations in order to check for the stability of the protein and ligand. Based on the RMSD plots presented in Figure 3A, that clearly showed that the conformation during production simulations was found to be fluctuating from 3.0 to 8.0 Å in Run 1 and 4.0 – 10.0 Å in Run 2. Run 1 was equilibrated well within 100 ns. For Run 2, RMSD started to equilibrate after 80 ns. This observation was explored through the root mean square fluctuations (RMSF) to determine which protein regions display higher levels of flexibility. Overall, the flexible regions were mainly found between residues ALA333 to PHE 344, and this region corresponded to the CPE12 peptide whereas the Claudin-4 regions were stable along the simulation time as expected (see Figure 3B). The hydrogen bonds formed in Claudin‑4 and CPE12 complex along the MD simulation were also analyzed to study the interaction between them as illustrated in Figure 4. The hydrogen bonds formed were found to be stable throughout the simulation.
Cloning of pET28a-cpe12-gfp
In order to develop the oral vaccine system via M cells targeting peptide, CPE12 was predicted in silico by bioinformatics methods as described above. Then it was produced in a large scale for in vitro and ex vivo experiments. For that reason, the recombinant peptide CPE12 was expressed in E. coli host strain. The construction of expression vector pET28a-cpe12-gfp was described in detail in “Materials and Methods” section. Gene cpe12-gfp was amplified from pET28a-gfp template (lane 2, Figure 5A). The fused gene gfp in C‑terminus, which is expressed to Green Fluorescent Protein, acts as a visual marker for assessing the interaction between targeting peptide and M cells. As for CPE12-GFP purification using nickel affinity chromatography, 6‑histidine residues were designed at the C-terminus next to the coding gene for GFP. Positive transformants containing clones cpe12 fused gfp were selected, screened (lane 2-3, Figure 5B) and sequenced (data not shown). As a result, cpe12 was cloned in frame with gfp.
Expression and purification of CPE12-GFP
Plasmid pET28a-cpe12-gfp was extracted, introduced into E. coli BL21 (DE3) and induced with IPTG for the protein expression. GFP was used as a control. As shown in Figure 3, an overexpressed protein band was roughly 30 kDa corresponding to GFP (lane 3, Figure 6A). Because CPE12 has only 12 amino acids, there was no significant difference in molecular weight comparing to GFP (lane 2, Figure 6A). In regard to solubility, it revealed that predictive protein CPE12-GFP was expressed in a soluble form (land 4-5, Figure 6A). CPE12‑GFP was designed to fuse with 6xHis tag, thus the expression of CPE12‑GFP could be confirmed by Western Blot probed with the specific anti-His antibody. The signals showed there were protein bands corresponding to GFP and CPE12‑GFP in total, soluble and insoluble forms located at the same size as SDS-PAGE result.
The soluble fraction was obtained as described in “Materials and Methods” section. CPE12-GFP in a soluble form was bound to the column (lane 1, Figure 6B). Unbound proteins flowed through the column (lane 2, Figure 6B). Residual contaminants and unbound proteins were removed with additional wash step of buffer B (lane 3-6, Figure 6B). Target protein CPE12-GFP was totally eluted with buffer C (lane 7-8, Figure 6B). Consequently, the purity of CPE12 was approximately 94.12%.
Ex vivo evaluation by M cells of mouse intestines
Expectedly, the recombinant CPE30-GFP (green) bound in a round shape on the lower part of the cell, not on the M cell surface, which was identified by binding with M cell-specific antibody, NKM 16-2-4 (purple) and rhodamine-conjugated UEA-1 (red) (Figure 7). In comparison to CPE30‑GFP, CPE12-GFP also bound to the lower part of the cell. In addition, the binding specificity of CPE12‑GFP to M cells is somewhat lower than that of CPE30-GFP.
DISCUSSION
In this study, we designed CPE12 peptide, derived from the C-terminal of 30 amino acids (CPE30) of Clostridium perfringens to target Claudin-4 using a combination of molecular modeling, docking, and molecular dynamics (MD) simulations. The stability of the docking complex in an aqueous environment was confirmed using molecular dynamics simulations. Trajectories obtained from the MD simulations were analyzed for equilibration that can be observed from the convergence of the RMSD plot, consisting RMSD of all Cα, N, and O atoms from each snapshot using the initial energy minimized structure as a reference. It is evident from Figure 3A that the simulation attained plateau and achieved convergence during 100 ns in Run 1. RMSF was performed to observe the flexibility of the complex. The RMSF profile of the CPE12 (short peptide) shows higher fluctuation as it becomes very flexible in solution. To investigate the protein-peptide interactions, binding free energy calculations were performed on 12-residue peptide CPE12 derived from the native CPE segment. To further understand the binding energetics of the complex, the MM-GBSA method was applied for binding free energy calculation (Table 1) The binding free energy of CPE12‑Claudin‑4 complex was -51.49 ± 0.37 kcal/mol in the average between two simulations run. The negative number indicated favorable binding. It was observed that the major favorable component contributing towards the binding of CPE12 and Claudin-4 were van der Waals in the gas phase, (∆Evdw = -55.92 ± 0.29 kcal/mol) and the non-polar part of the free energy of solvation (ESURF = ‑9.37 ± 0.22 kcal/mol). These two terms resulted in the total favorable non-polar interactions of -65.29 ± 0.36 kcal/mol that might come from hydrophobic interactions of CPE12 and Claudin-4. On the hand, favorable coulombic interactions (EEL = -68.75 ± 0.89 kcal/mol) were observed. To investigate the contributions of individual residues in total binding free energy, per residue free energy decomposition was used that also elucidate residue importance in interface binding. As shown in Figure 2, residues ILE196, VAL197, THR198, VAL211, LYS221, VAL300 belonging to Claudin-4 and TYR335, ILE339, IEU340, PHE341, PHE344 belonging to CPE12 were found to contribute most to the binding energy with the absolute relative decomposition energy greater than 1 kcal/mol. MD simulation suggests that CPE12 was stable in the binding pocket of Claudin-4 with observed hydrogen bond throughout the 100 ns MD simulations.
As mentioned above, in order to facilitate later observations of the interaction between CPE12 and its receptor on M cell surface visually, Green Fluorescent Protein was fused to CPE12 because of its fluorescent emission. In addition, the recombinant protein CPE12-GFP was directly purified through His‑IMAC method with the purity up to 94.12%, which was fully permitted to the next experiments as an ex vivo test on murine M cell.
CONCLUSION
In summary, the binding structure and its interaction of the peptide, CPE12 derived from the C‑terminal of CPE30 of Clostridium perfringens, were predicted by molecular docking and molecular dynamics simulations. MD simulation suggested that CPE12 had the ability to stably bind to M cell receptor Claudin‑4. In particular, CPE12 was proven that it had an adequate binding to Claudin-4 comparing to CPE30 via in vitro and ex vivo tests on murine M cells. This study laid the groundwork from in silico to ex vivo study for bioinformatics screening using molecular docking and molecular dynamics simulation to obtain the potent peptide targeting M cell.
ACKNOWLEDGEMENT
This research was funded by Vietnam National University HoChiMinh City (VNU-HCM) under Grant [C2018-18-06] and the University of Malaya under Grant [GPF063B-2018] through SATU co-PI program. Dr. Y.-S. Jang was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2017R1A6A1A03015876).
REFERENCES
- Bottalico L, Castellaneta F, Charitos IA. From Hydrotherapy to The Discovery of The Gut Microbiota: The Historical Gastrointestinal Health Concept. Pharmacophores. 2020;11(2):82-90.
- Alsimail MW, Alnaim AA, Alramadhan FS, Sagga BK, Alnomari LF, Almeashi NA, Jambi SK, Alaithan FA, Alshoura RA, Alotaibe NN, Alshalawi AA. Role of Gastrografin Challenge in Diagnosis of Small Intestinal Obstruction. Arch of Pharm Pract. 2019;10(4):21-5.
- Dika–Haxhirexha F, Santacroce L, Topi S, Alimani-Jakupi J, Haxhirexha A. Intestinal Parasitosis In Children: A Balkan Pilot Study. Pharmacophores. 2020;11(2):91-4.
- Jafar M, Khalid MS, Alghamdi HA, Al-Qurain EA, Alalwan ZH, Alshawwaf SA, Al Mahfoudh ZA, Al-Darwiesh AN. Improved Analgesic Effect and Alleviated Gastric Problems of Diclofenac Sodium Through a Gastric Floating in-Situ Gelling System. Int. J. Pharm. Phytopharm. Res. 2018;8(4):42-52.
- Wang M, Gao Z, Zhang Z, Pan L, Zhang Y. Roles of M cells in infection and mucosal vaccines. Human vaccines & immunotherapeutics. 2014 Dec 2;10(12):3544-51.
- Neutra MR, Pringault E, Kraehenbuhl JP. Antigen sampling across epithelial barriers and induction of mucosal immune responses. Annual review of immunology. 1996 Apr;14(1):275-300.
- Kunisawa J, Kurashima Y, Kiyono H. Gut-associated lymphoid tissues for the development of oral vaccines. Advanced drug delivery reviews. 2012 May 1;64(6):523-30.
- Hase K, Kawano K, Nochi T, Pontes GS, Fukuda S, Ebisawa M, Kadokura K, Tobe T, Fujimura Y, Kawano S, Yabashi A. Uptake through glycoprotein 2 of FimH+ bacteria by M cells initiates mucosal immune response. Nature. 2009 Nov;462(7270):226-30.
- Hoa, N. T. T., Truong, D. T., Hieu, T. V. Cloning and expression of GFP-fused m cell targeting protein (FimH), International Symposium on Green Technology (ISGT), 2016; 1009-1017.
- Kim SH, Jung DI, Yang IY, Kim J, Lee KY, Nochi T, Kiyono H, Jang YS. M cells expressing the complement C5a receptor are efficient targets for mucosal vaccine delivery. European journal of immunology. 2011 Nov;41(11):3219-29.
- An NH, Truong DT, Van Hieu T. Cloning, expression and purification of m-cell specific binding peptide (CO1) fused with GFP. Vietnam Journal of Biotechnology. 2016;14(4):599-604.
- Kim SH, Seo KW, Kim J, Lee KY, Jang YS. The M cell-targeting ligand promotes antigen delivery and induces antigen-specific immune responses in mucosal vaccination. The Journal of Immunology. 2010 Nov 15;185(10):5787-95.
- Freedman JC, Shrestha A, McClane BA. Clostridium perfringens enterotoxin: action, genetics, and translational applications. Toxins. 2016 Mar;8(3):73.
- Lo DD, Ling J, Eckelhoefer AH. M cell targeting by a Claudin 4 targeting peptide can enhance mucosal IgA responses. BMC biotechnology. 2012 Dec;12(1):1-9.
- Ling J, Liao H, Clark R, Wong MS, Lo DD. Structural constraints for the binding of short peptides to claudin-4 revealed by surface plasmon resonance. Journal of Biological Chemistry. 2008 Nov 7;283(45):30585-95.
- Shinoda T, Shinya N, Ito K, Ohsawa N, Terada T, Hirata K, Kawano Y, Yamamoto M, Kimura-Someya T, Yokoyama S, Shirouzu M. Structural basis for disruption of claudin assembly in tight junctions by an enterotoxin. Scientific reports. 2016 Sep 20;6(1):1-0.
- Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic acids research. 2004 Jul 1;32(suppl_2):W665-7.
- Van Zundert GC, Rodrigues JP, Trellet M, Schmitz C, Kastritis PL, Karaca E, Melquiond AS, van Dijk M, De Vries SJ, Bonvin AM. The HADDOCK2. 2 web server: user-friendly integrative modeling of biomolecular complexes. Journal of molecular biology. 2016 Feb 22;428(4):720-5.
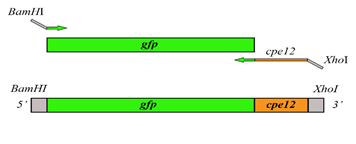
Figure 1: The schematic outline for the cloning of pET28a-cpe12-gfp.
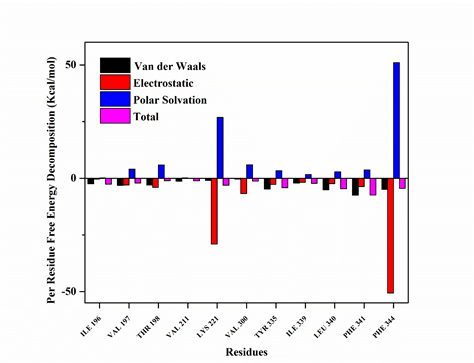
Figure 2: MM-GBSA decomposition of binding energy on a per-residue basis, only residues making significant favorable contributions are shown. For per residue decomposition analysis, 100 frames were extracted from the last 3 ns.
Figure 3: Root mean square deviation (RMSD) of the Cα atoms as a function of the MD simulation time (A) Root-mean-square fluctuation (RMSF) of the Cα atoms versus residue number (B).
Figure 4: The number of hydrogen bonds formed between Claudin-4 and CPE12 peptides during a 100 ns MD simulation.
Figure 5: Construction of plasmid pET-cpe12-gfp.
(A) Amplification of gene cpe12-gfp. M, DNA ladder; 1, negative control; 2, gene cpe12‑gfp. Gene cpe12 fuse gfp was designed at the size 754 bp.
(B) Screening of E. coli DH5α cells carrying plasmid pET-cpe12-gfp. M, DNA ladder; 1, negative control; 2-3, candidate transformants.
Figure 6: Expression and purification of CPE12-GFP.
(A) Identification of CPE12-GFP expression with SDS-PAGE and Western Blot. M, protein ladder; 1, negative control; 2, E. coli BL21 (DE3) pET28a-gfp in total; 3-5, E. coli BL21 (DE3)/pET2-cpe12-gfp in total, supernatant and precipitation, respectively.
(B) Analysis of purified fractions with SDS-PAGE and silver staining. M, protein ladder; 1, soluble protein fraction; 2, flow-through fraction; 3-6, fractions washed with buffer B; 7-8, eluted fractions with buffer C.
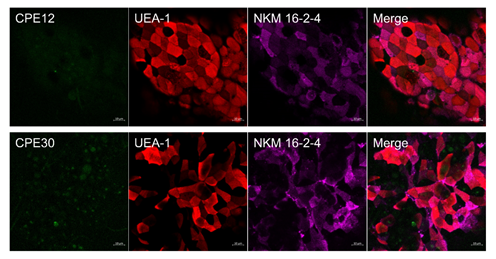
Figure 7: Ex vivo binding assay of CPEs to murine intestinal M cells.
Murine Peyer’s patches were initially fixed and permeabilized. CPE-GFPs were incubated with the fixed Peyer’s patches. Samples were then incubated with M cell-specific Abs NKM 16-2-4 (purple) and UEA-1-Rhodamine (red) to locate murine M cells. Scale bar: 10 mm.
Table 1: Binding Free Energy of CPE12/Claudin 4 complex and its components calculated using the MM-GBSA method.
|
Energy components |
MM-GBSA (kcal/mol) |
Standard Error of Mean |
|
van der Waals |
-68.91 |
0.21 |
|
EEL |
-43.32 |
0 .66 |
|
EGB |
80.64 |
0.60 |
|
ESURF |
-7.46 |
0.01 |
|
Delta G Gas |
-112.23 |
0.68 |
|
Delta G Sol |
73.18 |
0.60 |
|
Delta Total |
-39.06 |
0.19 |
The contributions are further broken into Ggas and Gsolv. The polar and non-polar contributions are EGB (or EPB) and ESURF (or ECAVITY/ENPOLAR), respectively for GB (or PB) calculations.
